- Details
- Written by: Emanuele Coccia
- Category: Research
- Hits: 2936
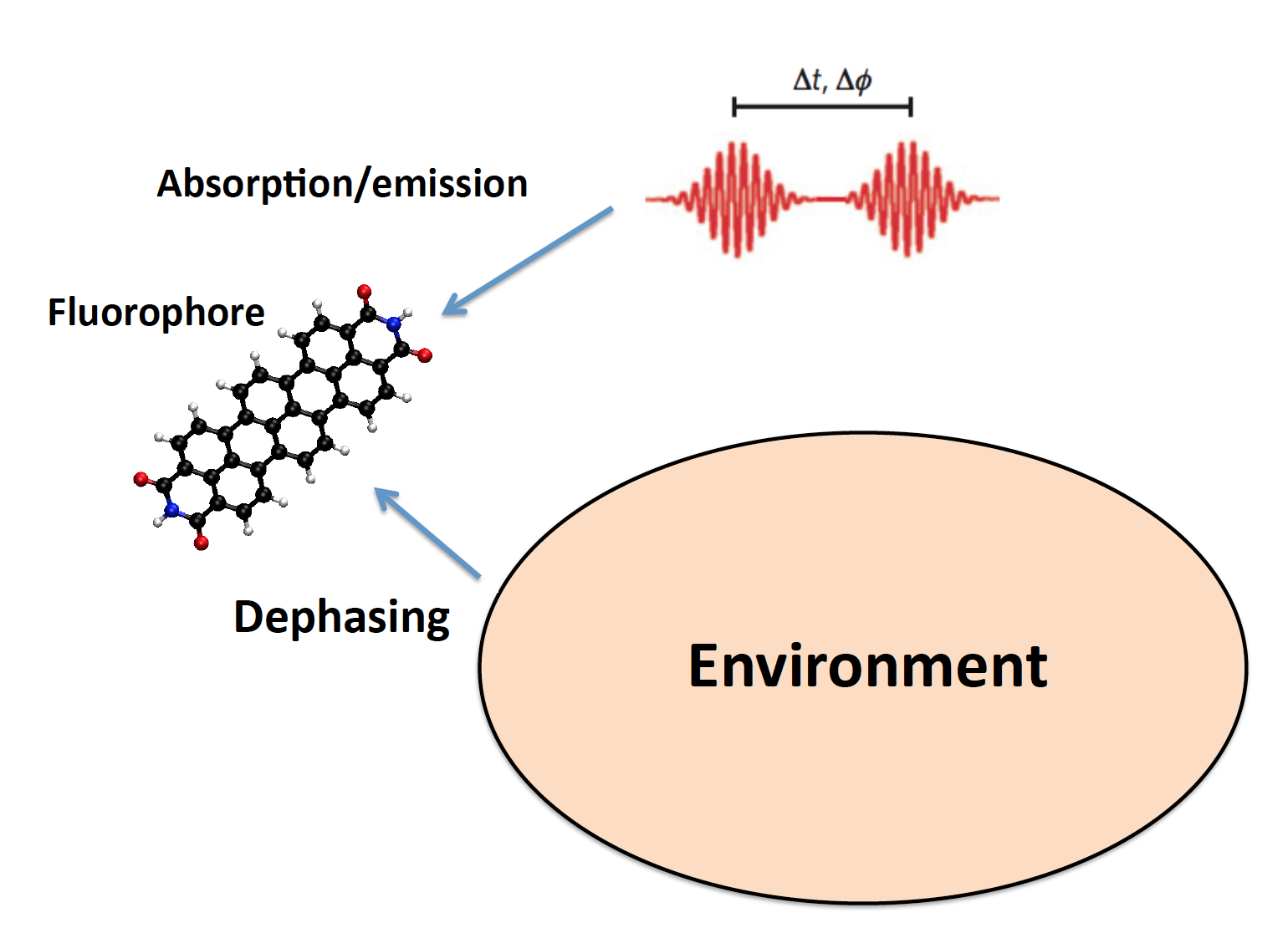
Ultrafast spectroscopy is a powerful tool to investigate, control and manipulate quantum coherence in molecules and complex systems. Detection of electronic and vibrational coherence in biological systems and in plasmonic nanostructures is a matter of stimulating and open debate. Recently, specific ultrafast spectroscopy techniques could probe a single molecule. To understand the outcomes of such experiments, theoretical and computational approaches are required, able to include all the important features of the simulated system. Using a recently developed computational approach coupling the theory of open quantum systems, by means of stochastic Schrödinger equation, with a quantum-chemical description of the molecular target, we have simulated a two-pulse experiment on organic fluorophores. Moreover, electronic decoherence can be thought as a further design element in molecular nanoplasmonics: metal nanoparticle effects on the absorption of light by a nearby molecule may be strongly affected (even qualitatively, i.e., suppression vs enhancement) by molecular decoherence. We are interested in developing time-resolved ab initio approaches to study electron dynamics in molecules and complex systems in presence of decoherence sources. (Emanuele Coccia)
- Details
- Written by: Emanuele Coccia
- Category: Research
- Hits: 1737
The Near Edge X-ray Absorption Fine Structure spectroscopy (NEXAFS) of molecules adsorbed on surfaces is widely used to investigate the orientation and geometry of the molecular adsorbate as well as the extent of the adsorbate-substrate interaction. The simulation of NEXAFS spectra of these systems represents a computational challenge for both a proper modelling of the adsorbate/surface system as well as for its size which can prevent the application of very accurate theoretical methods. We calculate the NEXAFS spectra in terms of excitation energies and oscillator strength values calculated at DFT or TDDFT level employing a cluster approach to mimic the adsorbate systems. Finite size cluster model for surface adsorption has proven very efficient and accurate to simulate NEXAFS spectra due to the localized nature of the core excitation process. Total spectra as well as angle resolved spectra can be simulated. Previous optimization of the surface and of the molecules adsorbed on it is performed by a periodic slab methodology in the frame of density functional theory (DFT). (Giovanna Fronzoni and Daniele Toffoli)
- Details
- Written by: Emanuele Coccia
- Category: Research
- Hits: 2408
Development of new methods for DFT and TDDFT
In our group we have a long collaboration with SCM (Amsterdam) since we are developers of the ADF (now AMS) suite of programs. In particular in ADF we have implemented core electron excitations, the polTDDFT method to calculate the optical properties of very large systems, and the Hybrid Diagonal Approximation (HDA) to speedup TDDFT calculations with hybrid kernels and Slater Type Orbitals (STO) basis sets. We also worked on analysis tools for TDDFT, as the fragment analysis and the ICM-OS.
- Theoretical study of metal clusters.
We are in particular interested in the optical properties of metal clusters, such as plasmonic behaviors, circular dichroism, rebirth of plasmons thanks to ligand effects and alloying effects.
(Mauro Stener and Daniele Toffoli)
- Details
- Written by: Emanuele Coccia
- Category: Research
- Hits: 2635

High-harmonic generation (HHG) is a highly nonlinear optical process induced by an intense infrared pulse. The atomic or molecular target emits high-order harmonics (i.e., integer multiples of the pulse frequency) up to X-ray region. HHG spectroscopy studies how intensity and shape of harmonic peaks encode information on electronic structure and dynamics of the target. We simulate HHG by means of time-dependent Schrödinger equation: the electronic wavepacket is propagated under the influence of a strong electric field with an intensity of the order of 1014 W/cm2 . The wavepacket is composed of field-free eigenstates computed at configuration-interaction or TDDFT level. We develop and apply this protocol to investigate the strong-field electron dynamics in femto and attosecond regime in molecules, as uracil (shown in the Figure). (Emanuele Coccia)
- Details
- Written by: Emanuele Coccia
- Category: Research
- Hits: 5200
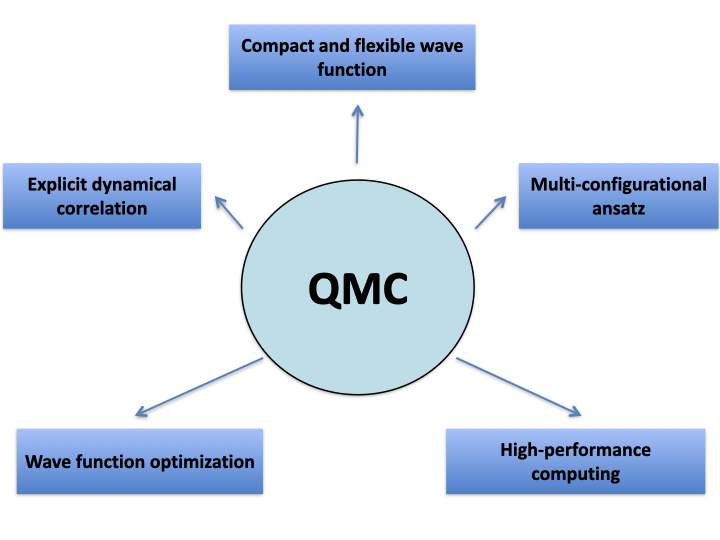
Quantum Monte Carlo (QMC) are the gold standard for correlated wavefunctions. Dynamical electron correlation is explicitly included in the QMC ansatz via the Jastrow factor, which depends on interparticle distances. QMC methods are embarrassing parallel, and allow us to efficiently exploit High-Performance Computing. We are interested in developing and applying QMC methods to electronic excited-state properties by means of linear-response theory. (Emanuele Coccia)